What is the disease all about?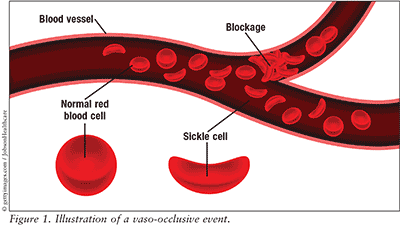
Sickle cell disease is an inherited blood disorder. It is marked by flawed hemoglobin. That’s the protein in red blood cells that carries oxygen to the tissues of the body. So, sickle cell disease interferes with the delivery of oxygen to the tissues.
Signs and symptoms of sickle cell disease usually begin in early childhood. Characteristic features of this disorder include a low number of red blood cells (anemia), repeated infections, and periodic episodes of pain. The severity of symptoms varies from person to person. Some people have mild symptoms, while others are frequently hospitalized for more serious complications.
Red blood cells with normal hemoglobin are smooth, disk-shaped, and flexible, like doughnuts without holes. They can move through the blood vessels easily. Cells with sickle cell hemoglobin are stiff and sticky. When they lose their oxygen, they form into the shape of a sickle or crescent, like the letter C. These cells stick together and can’t easily move through the blood vessels. This can block small blood vessels and the movement of healthy, normal oxygen-carrying blood. The blockage can cause pain.
Impact of the disease on individuals and society
Sickle Cell Disorder is a global health problem with psychosocial implications. Nigeria has the largest population of people with sickle cell disorder, with about 150,000 births annually. This study explored the psychosocial impact of sickle cell disorder in 408 adolescents and adults attending three hospitals in Lagos, Nigeria.
Some issues in education, employment, and healthcare were expressed, however these were in the minority of cases. The results also showed that depressive feelings were experienced in almost half the study population, even though feelings of anxiety or self-hate were uncommon. Rarely, extreme conditions such as severe dehydration and high-intensity physical activity can lead to serious health issues, including sudden death, in individuals with sickle cell trait.
What techniques can be used to diagnose the disease?
Early diagnosis and prevention of complications is critical in sickle cell disease treatment. Treatment aims to prevent organ damage including strokes, prevent infection, and treat symptoms. Treatment may include:
- Pain medications. This is for sickle cell crises.
- Drinking plenty of water daily (8 to 10 glasses). This is to prevent and treat pain crises. In some situations, intravenous fluids may be required.
- Blood transfusions. These may help treat anemia and prevent stroke. They are also used to dilute the sickled hemoglobin with normal hemoglobin to treat chronic pain, acute chest syndrome, splenic sequestration, and other emergencies.
- Vaccinations and antibiotics. These are used to prevent infections.
- Folic acid. Folic acid will help prevent severe anemia.
- Hydroxyurea. This medication helps reduce the frequency of pain crises and acute chest syndrome. It may also help decrease the need for blood transfusions. The long-term effects of the medication are unknown.
- Regular eye exams. These are done to screen for retinopathy.
- Bone marrow transplant. Bone marrow transplants can cure some people with sickle cell disease. The decision to have this procedure is based on the severity of the disease and ability to find a suitable bone marrow donor. These decisions need to be discussed with your doctor and are only done at specialized medical centers.
Which other systems could be affected?
Any and all major organs are affected by sickle cell disease. The liver, heart, kidneys, gallbladder, eyes, bones, and joints can suffer damage from the abnormal function of the sickle cells and their inability to flow through the small blood vessels correctly.
How can transplantation or any other scientific advances play an important role in the treatment of the disease?
A bone marrow transplant replaces the cells in your body that make red blood cells, called hematopoietic stem cells, with new ones. That means your body will stop making the sickle-shaped cells that cause the disease.

ETHICAL ISSUES
The issue of safety of the procedures used for PND is worth mentioning first. Though they are found to be relatively safe, there is still a chance of a miscarriage following chorionic villus sampling and amniocentesis
Abortion of the affected fetus is regarded as a component of PND in most cases. In the case of the fetus having the SS genotype, the ethical question arises whether to have an abortion or to keep the pregnancy. The decision whether to terminate a pregnancy based on a positive result is usually a difficult one that involves religious, psychosocial and cultural considerations .Utilitarians would argue that it is more cost effective to abort the affected fetuses as this will reduce on the long run the socio-economic and emotional consequences of the disease. Bringing up the question of cost-effectiveness and life of patients would be looked at in many developing countries like Nigeria as being cold and inhuman, but the reality of scarcity of resources and rationing of health care resources is there for all to see.
SOCIAL ISSUES :
Sickle cell disease (SCD) in Britain affects some 150 babies a year, born mainly to Afro-Caribbean and West African families. Like every chronic illness, it can have major psychosocial and practical consequences. Kenny Midence and Polly Shand describe the condition and the problems facing people with SCD and their families.
CULTURAL ISSUES :
The strongest support exists for claims of social deficits among adolescents and depression and work-related problems among adults. The social context of SCD, including issues related to socioeconomic status (SES), urbanicity, ethnicity, cultural values, and racial stigmatization, are important to include in empirical assessments and theoretical analyses of the effects of SCD on children and their families. The adverse psychosocial functioning often described as an effect of SCD might indeed be a consequence of these factors acting alone or in concert with the strains of SCD.